---
title: AET06
subtitle: Adverse Events by Baseline Characteristic
---
------------------------------------------------------------------------
{{< include ../../_utils/envir_hook.qmd >}}
```{r setup, echo = FALSE, warning = FALSE, message = FALSE}
library(dplyr)
library(tern)
adsl <- random.cdisc.data::cadsl
adae <- random.cdisc.data::cadae
adsub <- random.cdisc.data::cadsub
adsub_bmi <- adsub %>%
filter(PARAMCD == "BBMISI") %>%
select(STUDYID, USUBJID, AVALCAT1) %>%
mutate(
AVALCAT1 = factor(AVALCAT1, levels = c("<18.5", "18.5 - 24.9", "25 - 29.9", ">30"))
)
adsl <- adsl %>%
mutate(
RACE1 = case_when(
RACE == "WHITE" ~ "WHITE",
TRUE ~ "NON-WHITE"
),
RACE1 = factor(
RACE1,
levels = c("WHITE", "NON-WHITE")
)
) %>%
left_join(
y = adsub_bmi,
by = c("STUDYID", "USUBJID")
)
adae_labels <- var_labels(adae)
adae <- adae %>%
mutate(
RACE1 = case_when(
RACE == "WHITE" ~ "WHITE",
TRUE ~ "NON-WHITE"
),
RACE1 = factor(
RACE1,
levels = c("WHITE", "NON-WHITE")
)
) %>%
left_join(
y = adsub_bmi,
by = c("STUDYID", "USUBJID")
)
var_labels(adae) <- c(adae_labels, "RACE1" = "RACE WHITE/NON-WHITE", "AVALCAT1" = "Baseline BMI Category")
# Ensure character variables are converted to factors and empty strings and NAs are explicit missing levels.
adsl <- df_explicit_na(adsl)
adae <- df_explicit_na(adae)
adsub_bmi <- df_explicit_na(adsub_bmi)
```
```{r include = FALSE}
webr_code_labels <- c("setup")
```
{{< include ../../_utils/webr_no_include.qmd >}}
## Output
:::::::: panel-tabset
## Adverse Events by Sex
::: {.panel-tabset .nav-justified group="webr"}
## {{< fa regular file-lines sm fw >}} Preview
```{r variant1, test = list(result_v1 = "result")}
split_fun <- drop_split_levels
lyt <- basic_table(show_colcounts = TRUE) %>%
split_cols_by("ARM") %>%
split_cols_by("SEX") %>%
analyze_num_patients(
vars = "USUBJID",
.stats = c("unique", "nonunique"),
.labels = c(
unique = "Total number of patients with at least one adverse event",
nonunique = "Overall total number of events"
)
) %>%
split_rows_by(
"AEBODSYS",
child_labels = "visible",
nested = FALSE,
split_fun = split_fun,
label_pos = "topleft",
split_label = obj_label(adae$AEBODSYS)
) %>%
summarize_num_patients(
var = "USUBJID",
.stats = c("unique", "nonunique"),
.labels = c(
unique = "Total number of patients with at least one adverse event",
nonunique = "Total number of events"
)
) %>%
count_occurrences(vars = "AEDECOD", .indent_mods = -1L) %>%
append_varlabels(adae, "AEDECOD", indent = 1L)
result <- build_table(
lyt = lyt,
df = adae,
alt_counts_df = adsl
) %>%
prune_table() %>%
# Sorted by decreasing frequency across all groups by System Organ Class and Preferred Term.
sort_at_path(
path = c("AEBODSYS"),
scorefun = cont_n_allcols
) %>%
sort_at_path(
path = c("AEBODSYS", "*", "AEDECOD"),
scorefun = score_occurrences
)
result
```
```{r include = FALSE}
webr_code_labels <- c("variant1")
```
{{< include ../../_utils/webr.qmd >}}
:::
## Adverse Events by Other Baseline <br/> Characteristics (e.g. Biomarker Group)
::: {.panel-tabset .nav-justified group="webr"}
## {{< fa regular file-lines sm fw >}} Preview
```{r variant2, test = list(result_v2 = "result")}
split_fun <- drop_split_levels
lyt <- basic_table(show_colcounts = TRUE) %>%
split_cols_by("ARM") %>%
split_cols_by("BMRKR2") %>%
analyze_num_patients(
vars = "USUBJID",
.stats = c("unique", "nonunique"),
.labels = c(
unique = "Total number of patients with at least one adverse event",
nonunique = "Overall total number of events"
)
) %>%
split_rows_by(
"AEBODSYS",
child_labels = "visible",
nested = FALSE,
split_fun = split_fun,
label_pos = "topleft",
split_label = obj_label(adae$AEBODSYS)
) %>%
summarize_num_patients(
var = "USUBJID",
.stats = c("unique", "nonunique"),
.labels = c(
unique = "Total number of patients with at least one adverse event",
nonunique = "Total number of events"
)
) %>%
count_occurrences(vars = "AEDECOD", .indent_mods = -1L) %>%
append_varlabels(adae, "AEDECOD", indent = 1L)
result <- build_table(
lyt = lyt,
df = adae,
alt_counts_df = adsl
) %>%
prune_table() %>%
# Sorted by decreasing frequency across all groups by System Organ Class and Preferred Term.
sort_at_path(
path = c("AEBODSYS"),
scorefun = cont_n_allcols
) %>%
sort_at_path(
path = c("AEBODSYS", "*", "AEDECOD"),
scorefun = score_occurrences
)
result
```
```{r include = FALSE}
webr_code_labels <- c("variant2")
```
{{< include ../../_utils/webr.qmd >}}
:::
## Adverse Events by Baseline Characteristic <br/> from ADSUB (e.g. BMI Category)
::: {.panel-tabset .nav-justified group="webr"}
## {{< fa regular file-lines sm fw >}} Preview
```{r variant3, test = list(result_v3 = "result")}
split_fun <- drop_split_levels
lyt <- basic_table(show_colcounts = TRUE) %>%
split_cols_by("ARM") %>%
split_cols_by("AVALCAT1") %>%
analyze_num_patients(
vars = "USUBJID",
.stats = c("unique", "nonunique"),
.labels = c(
unique = "Total number of patients with at least one adverse event",
nonunique = "Overall total number of events"
)
) %>%
split_rows_by(
"AEBODSYS",
child_labels = "visible",
nested = FALSE,
split_fun = split_fun,
label_pos = "topleft",
split_label = obj_label(adae$AEBODSYS)
) %>%
summarize_num_patients(
var = "USUBJID",
.stats = c("unique", "nonunique"),
.labels = c(
unique = "Total number of patients with at least one adverse event",
nonunique = "Total number of events"
)
) %>%
count_occurrences(vars = "AEDECOD", .indent_mods = -1L) %>%
append_varlabels(adae, "AEDECOD", indent = 1L)
result <- build_table(
lyt = lyt,
df = adae,
alt_counts_df = adsl
) %>%
prune_table() %>%
# Sorted by decreasing frequency across all groups by System Organ Class and Preferred Term.
sort_at_path(
path = c("AEBODSYS"),
scorefun = cont_n_allcols
) %>%
sort_at_path(
path = c("AEBODSYS", "*", "AEDECOD"),
scorefun = score_occurrences
)
result
```
```{r include = FALSE}
webr_code_labels <- c("variant3")
```
{{< include ../../_utils/webr.qmd >}}
:::
## Adverse Events by Regrouped Baseline <br/> Characteristics (e.g. Race)
::: {.panel-tabset .nav-justified group="webr"}
## {{< fa regular file-lines sm fw >}} Preview
```{r variant4, test = list(result_v4 = "result")}
split_fun <- drop_split_levels
lyt <- basic_table(show_colcounts = TRUE) %>%
split_cols_by("ARM") %>%
split_cols_by("RACE1") %>%
analyze_num_patients(
vars = "USUBJID",
.stats = c("unique", "nonunique"),
.labels = c(
unique = "Total number of patients with at least one adverse event",
nonunique = "Overall total number of events"
)
) %>%
split_rows_by(
"AEBODSYS",
child_labels = "visible",
nested = FALSE,
split_fun = split_fun,
label_pos = "topleft",
split_label = obj_label(adae$AEBODSYS)
) %>%
summarize_num_patients(
var = "USUBJID",
.stats = c("unique", "nonunique"),
.labels = c(
unique = "Total number of patients with at least one adverse event",
nonunique = "Total number of events"
)
) %>%
count_occurrences(vars = "AEDECOD", .indent_mods = -1L) %>%
append_varlabels(adae, "AEDECOD", indent = 1L)
result <- build_table(
lyt = lyt,
df = adae,
alt_counts_df = adsl
) %>%
prune_table() %>%
prune_table() %>%
# Sorted by decreasing frequency across all groups by System Organ Class and Preferred Term.
sort_at_path(
path = c("AEBODSYS"),
scorefun = cont_n_allcols
) %>%
sort_at_path(
path = c("AEBODSYS", "*", "AEDECOD"),
scorefun = score_occurrences
)
result
```
```{r include = FALSE}
webr_code_labels <- c("variant4")
```
{{< include ../../_utils/webr.qmd >}}
:::
## Adverse Events by Baseline Characteristics <br/> (e.g. Sex) Including High-Level Terms
::: {.panel-tabset .nav-justified group="webr"}
## {{< fa regular file-lines sm fw >}} Preview
```{r variant5, test = list(result_v5 = "result")}
split_fun <- drop_split_levels
lyt <- basic_table(show_colcounts = TRUE) %>%
split_cols_by("ARM") %>%
split_cols_by("SEX") %>%
analyze_num_patients(
vars = "USUBJID",
.stats = c("unique", "nonunique"),
.labels = c(
unique = "Total number of patients with at least one adverse event",
nonunique = "Overall total number of events"
)
) %>%
split_rows_by(
"AEBODSYS",
child_labels = "visible",
nested = FALSE,
split_fun = split_fun,
label_pos = "topleft",
split_label = obj_label(adae$AEBODSYS)
) %>%
summarize_num_patients(
var = "USUBJID",
.stats = c("unique", "nonunique"),
.labels = c(
unique = "Total number of patients with at least one adverse event",
nonunique = "Total number of events"
)
) %>%
split_rows_by(
"AEHLT",
child_labels = "visible",
indent_mod = -1L,
split_fun = split_fun,
label_pos = "topleft",
split_label = obj_label(adae$AEHLT)
) %>%
summarize_num_patients(
var = "USUBJID",
.stats = c("unique", "nonunique"),
.labels = c(
unique = "Total number of patients with at least one adverse event",
nonunique = "Total number of events"
)
) %>%
count_occurrences("AEDECOD", .indent_mods = -1L) %>%
append_varlabels(adae, "AEDECOD", indent = 2L)
result <- build_table(
lyt = lyt,
df = adae,
alt_counts_df = adsl
) %>%
prune_table() %>%
# Sorted by decreasing frequency across all groups by System Organ Class, High-Level Term and Preferred Term.
sort_at_path(
path = c("AEBODSYS"),
scorefun = cont_n_allcols
) %>%
sort_at_path(
path = c("AEBODSYS", "*", "AEHLT"),
scorefun = cont_n_allcols
) %>%
sort_at_path(
path = c("AEBODSYS", "*", "AEHLT", "*", "AEDECOD"),
scorefun = score_occurrences
)
result
```
```{r include = FALSE}
webr_code_labels <- c("variant5")
```
{{< include ../../_utils/webr.qmd >}}
:::
## Data Setup
```{r setup}
#| code-fold: show
```
::::::::
{{< include ../../_utils/save_results.qmd >}}
## `teal` App
::: {.panel-tabset .nav-justified}
## {{< fa regular file-lines fa-sm fa-fw >}} Preview
```{r teal, opts.label = c("skip_if_testing", "app")}
library(teal.modules.clinical)
## Data reproducible code
data <- teal_data()
data <- within(data, {
ADSL <- random.cdisc.data::cadsl
ADAE <- random.cdisc.data::cadae
# Ensure character variables are converted to factors and empty strings and NAs are explicit missing levels.
ADSL <- df_explicit_na(ADSL)
ADAE <- df_explicit_na(ADAE)
})
join_keys(data) <- default_cdisc_join_keys[c("ADSL", "ADAE")]
## Reusable Configuration For Modules
ADAE <- data[["ADAE"]]
## Setup App
app <- init(
data = data,
modules = modules(
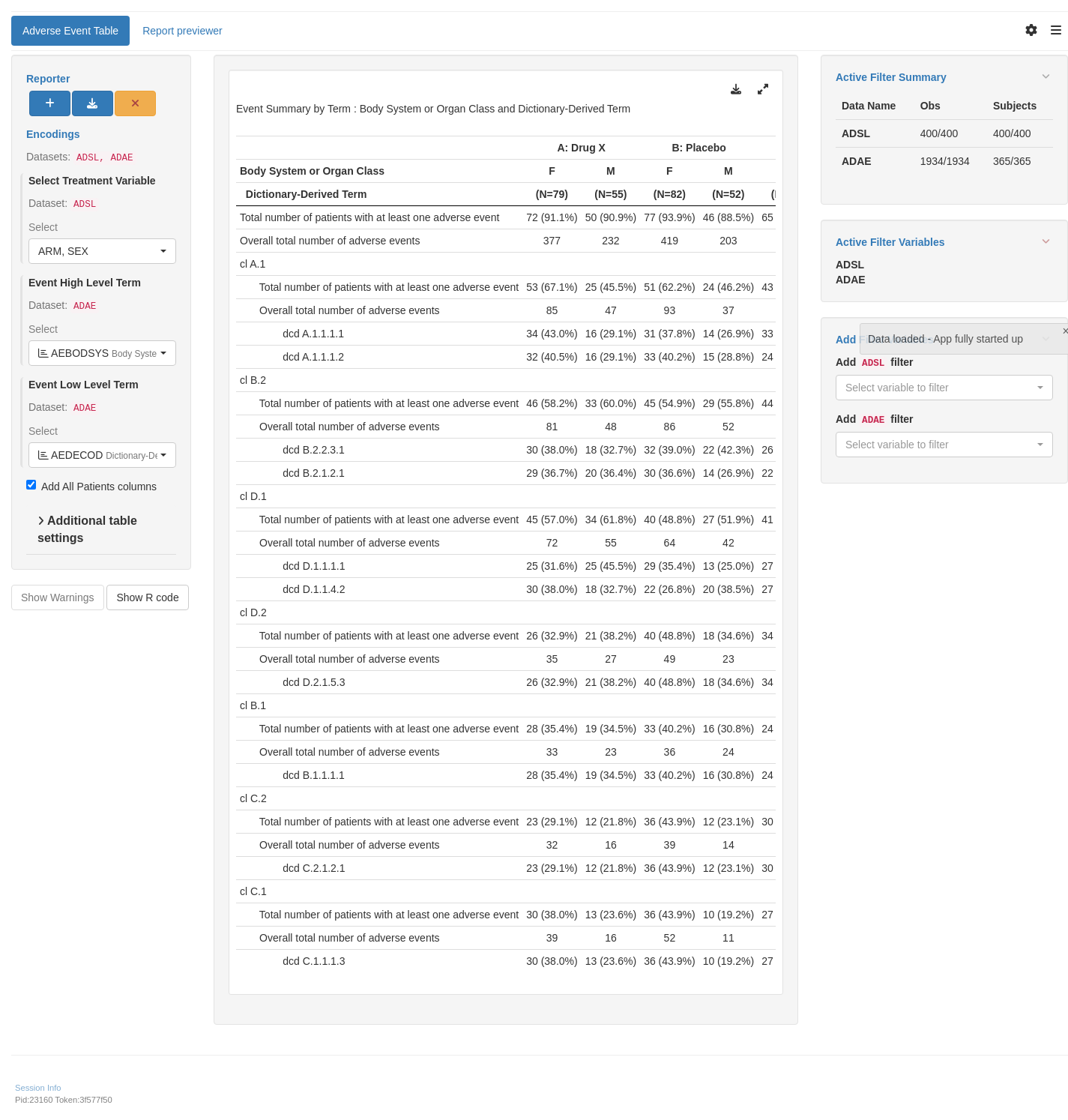
tm_t_events(
label = "Adverse Event Table",
dataname = "ADAE",
arm_var = choices_selected(c("ARM", "ARMCD", "SEX"), c("ARM", "SEX")),
llt = choices_selected(
choices = variable_choices(ADAE, c("AETERM", "AEDECOD")),
selected = c("AEDECOD")
),
hlt = choices_selected(
choices = variable_choices(ADAE, c("AEBODSYS", "AESOC")),
selected = "AEBODSYS"
),
add_total = TRUE,
event_type = "adverse event"
)
)
)
shinyApp(app$ui, app$server)
```
{{< include ../../_utils/shinylive.qmd >}}
:::
{{< include ../../repro.qmd >}}