RNAG9
RNAseq Kaplan-Meier Graph
This page can be used as a template of how to produce Kaplan-Meier graphs for RNA-seq gene expression analysis using available tern and hermes functions, and to create an interactive Kaplan-Meier graph for RNA-seq gene expression analysis using teal.modules.hermes.
The code below needs both RNA-seq data (in HermesData format) and time-to-event data (in ADTTE format) as input.
We first prepare the time-to-event data. We define an event indicator variable, transform the time to months and filter down to the overall survival subset.
Then we prepare the RNA-seq data. See RNAG1 for basic details on how to import, filter and normalize HermesData. We use col_data_with_genes() to extract the sample variables (colData) from the object, together with a single specified gene or a specified gene signature. See ?hermes::gene_spec for details on how to do this. Then we use inner_join_cdisc() to join this genetic data with the ADTTE data from above. See the help page for more details, in particular how the join keys can be customized if needed - here we just join based on USUBJID by default.
We can then cut the resulting gene column (we figure out the column name and save it in arm_name below) in the joined_data into quantile bins (in this example we want three equally sized groups).
It is now simple to create the Kaplan-Meier graph by providing the data set created above with the variable specification. Note that we specify the above created gene_factor as arm variable here.
Code
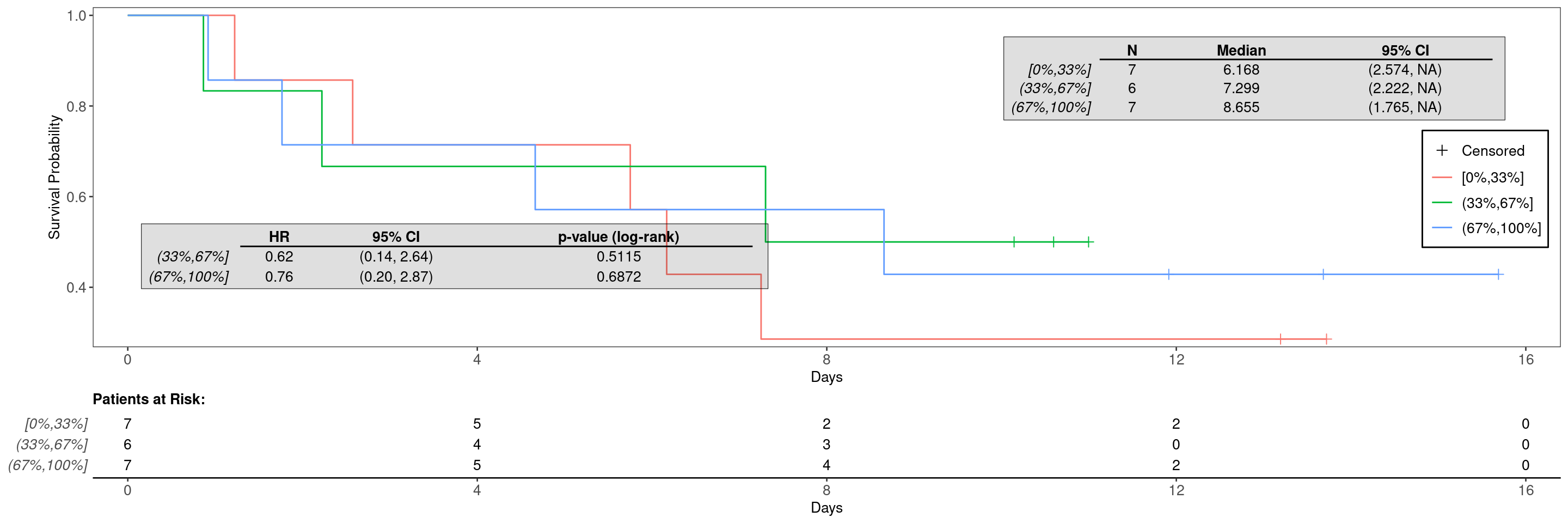
See KG1 to KG5 for additional customization options for the Kaplan-Meier graphs or the help page ?g_km().
We start by importing a MultiAssayExperiment and sample ADTTE data; here we use the example multi_assay_experiment available in hermes and example ADTTE data from random.cdisc.data. We can then use the provided teal module tm_g_km to include the corresponding interactive Kaplan-Meier analysis in our teal app. Note that by default the counts assay is excluded via the exclude_assays argument, but we can include it by just saying that we don’t want to exclude any assays. In case that we have different non-standard column names in our ADTTE data set we could also specify them via the adtte_vars argument, see the documentation ?tm_g_km for more details.
Code
Warning: `datanames<-()` was deprecated in teal.data 0.7.0.
ℹ invalid to use `datanames()<-` or `names()<-` on an object of class
`teal_data`. See ?names.teal_dataCode
Warning: 'experiments' dropped; see 'drops()'
R version 4.5.2 (2025-10-31)
Platform: x86_64-pc-linux-gnu
Running under: Ubuntu 24.04.4 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
time zone: Etc/UTC
tzcode source: system (glibc)
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] random.cdisc.data_0.3.16 teal.modules.hermes_0.2.0
[3] teal_1.1.0 teal.slice_0.7.1
[5] teal.data_0.8.0 teal.code_0.7.1
[7] shiny_1.13.0 hermes_1.14.0
[9] SummarizedExperiment_1.40.0 Biobase_2.70.0
[11] GenomicRanges_1.62.1 Seqinfo_1.0.0
[13] IRanges_2.44.0 S4Vectors_0.48.0
[15] BiocGenerics_0.56.0 generics_0.1.4
[17] MatrixGenerics_1.22.0 matrixStats_1.5.0
[19] ggfortify_0.4.19 ggplot2_4.0.2
[21] dplyr_1.2.1 tern_0.9.10
[23] rtables_0.6.15 magrittr_2.0.5
[25] formatters_0.5.12
loaded via a namespace (and not attached):
[1] RColorBrewer_1.1-3 jsonlite_2.0.0
[3] shape_1.4.6.1 nestcolor_0.1.3
[5] MultiAssayExperiment_1.36.2 farver_2.1.2
[7] rmarkdown_2.31 fs_2.0.1
[9] ragg_1.5.2 GlobalOptions_0.1.4
[11] vctrs_0.7.2 memoise_2.0.1
[13] webshot_0.5.5 htmltools_0.5.9
[15] S4Arrays_1.10.1 forcats_1.0.1
[17] BiocBaseUtils_1.13.0 progress_1.2.3
[19] curl_7.0.0 broom_1.0.12
[21] SparseArray_1.10.10 sass_0.4.10
[23] bslib_0.10.0 fontawesome_0.5.3
[25] htmlwidgets_1.6.4 httr2_1.2.2
[27] cachem_1.1.0 teal.widgets_0.6.0
[29] mime_0.13 lifecycle_1.0.5
[31] iterators_1.0.14 pkgconfig_2.0.3
[33] webshot2_0.1.2 Matrix_1.7-5
[35] R6_2.6.1 fastmap_1.2.0
[37] rbibutils_2.4.1 clue_0.3-68
[39] digest_0.6.39 colorspace_2.1-2
[41] shinycssloaders_1.1.0 ps_1.9.2
[43] AnnotationDbi_1.72.0 textshaping_1.0.5
[45] RSQLite_2.4.6 filelock_1.0.3
[47] labeling_0.4.3 httr_1.4.8
[49] abind_1.4-8 compiler_4.5.2
[51] bit64_4.6.0-1 withr_3.0.2
[53] doParallel_1.0.17 S7_0.2.1
[55] backports_1.5.1 bsicons_0.1.2
[57] DBI_1.3.0 logger_0.4.1
[59] biomaRt_2.66.2 rappdirs_0.3.4
[61] DelayedArray_0.36.1 rjson_0.2.23
[63] chromote_0.5.1 tools_4.5.2
[65] otel_0.2.0 httpuv_1.6.17
[67] glue_1.8.0 callr_3.7.6
[69] promises_1.5.0 grid_4.5.2
[71] checkmate_2.3.4 cluster_2.1.8.2
[73] gtable_0.3.6 websocket_1.4.4
[75] tidyr_1.3.2 hms_1.1.4
[77] XVector_0.50.0 ggrepel_0.9.8
[79] foreach_1.5.2 pillar_1.11.1
[81] stringr_1.6.0 later_1.4.8
[83] circlize_0.4.18 splines_4.5.2
[85] BiocFileCache_3.0.0 lattice_0.22-9
[87] survival_3.8-6 bit_4.6.0
[89] tidyselect_1.2.1 ComplexHeatmap_2.26.1
[91] Biostrings_2.78.0 knitr_1.51
[93] gridExtra_2.3 teal.logger_0.4.1
[95] xfun_0.57 stringi_1.8.7
[97] yaml_2.3.12 shinyWidgets_0.9.1
[99] evaluate_1.0.5 codetools_0.2-20
[101] tibble_3.3.1 cli_3.6.5
[103] systemfonts_1.3.2 xtable_1.8-8
[105] Rdpack_2.6.6 jquerylib_0.1.4
[107] processx_3.8.7 dichromat_2.0-0.1
[109] Rcpp_1.1.1 teal.reporter_0.6.1
[111] dbplyr_2.5.2 png_0.1-9
[113] parallel_4.5.2 assertthat_0.2.1
[115] blob_1.3.0 prettyunits_1.2.0
[117] scales_1.4.0 purrr_1.2.1
[119] crayon_1.5.3 GetoptLong_1.1.0
[121] rlang_1.2.0 formatR_1.14
[123] cowplot_1.2.0 KEGGREST_1.50.0
[125] shinyjs_2.1.1