We will use the cadtte data set from the random.cdisc.data package to create the STEP survival graph. We start by filtering the adtte data set for the overall survival observations, converting time of overall survival to months, creating a new variable for event information, binarizing the ARM variable, and discarding BMRKR1 values for the non-BEP. We also relabel the biomarker evaluable population flag variable BEP01FL.
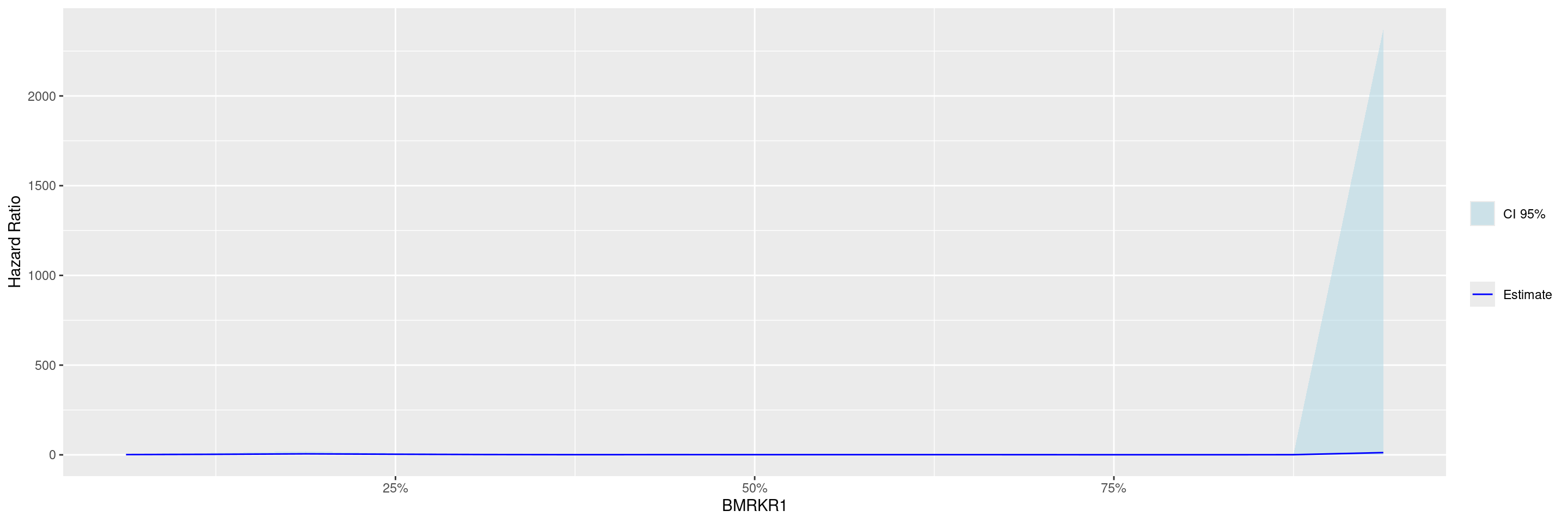
We then perform with the fit_survival_step() function the required calculations: this function fits the Subgroup Treatment Effect Pattern (STEP) models for the survival outcome within each of the percentile intervals of the biomarker variable defining the subgroups. The treatment arm variable must have exactly two levels, where the first one is taken as reference, i.e. the estimated hazard ratios are for the comparison of the second level vs. the first one.
In this example we fit the default model where a constant treatment effect is estimated in each of the subgroups that are created according to biomarker quantiles.
In this second example we fit instead a model with quadratic biomarker interaction term and we control the number of points at which the hazard ratios are estimated.
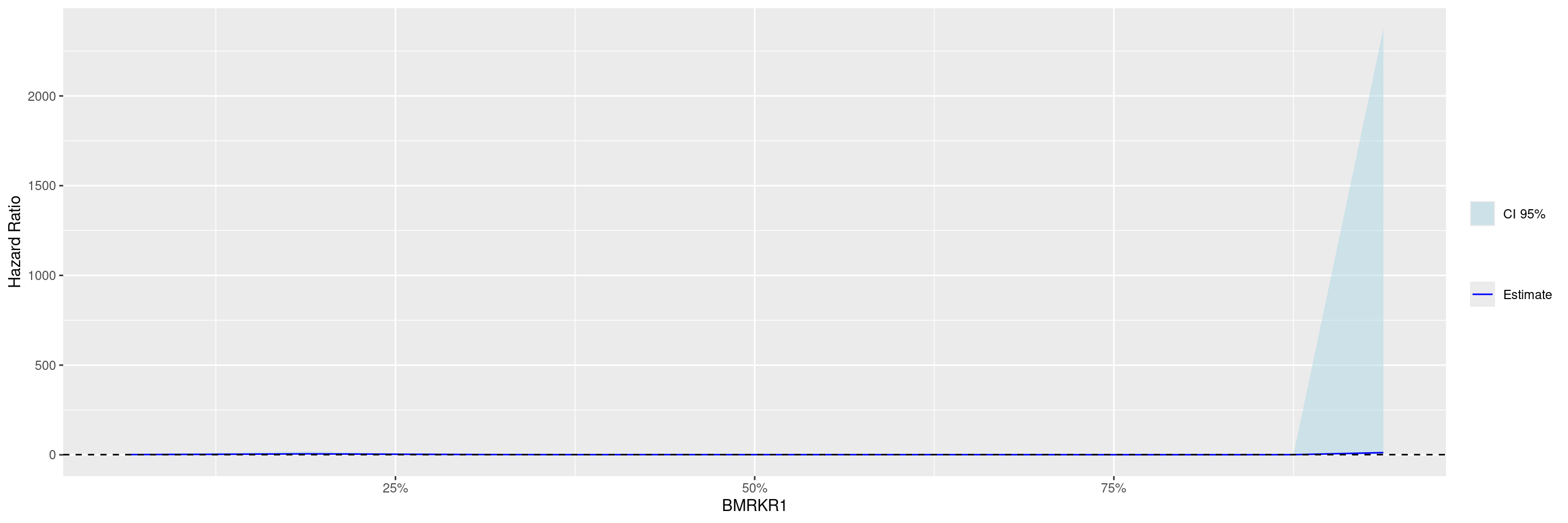
Before we can plot the data, the broom::tidy() method needs to be applied to the STEP result to obtain graph ready data. Thereafter with the g_step() function we can create a graph showing the estimated HR along the continuous biomarker subgroups.
---title: SPG1subtitle: STEP Graphs for Survival Outcomecategories: [SPG]---------------------------------------------------------------------------::: panel-tabset## SetupWe will use the `cadtte` data set from the `random.cdisc.data` package to create the STEP survival graph.We start by filtering the `adtte` data set for the overall survival observations, converting time of overall survival to months, creating a new variable for event information, binarizing the `ARM` variable, and discarding `BMRKR1` values for the non-BEP.We also relabel the biomarker evaluable population flag variable `BEP01FL`.```{r, message = FALSE}library(tern)library(ggplot2.utils)library(dplyr)adtte <- random.cdisc.data::cadtte %>% df_explicit_na() %>% filter( PARAMCD == "OS" ) %>% mutate( AVAL = day2month(AVAL), AVALU = "Months", is_event = CNSR == 0, ARM_BIN = fct_collapse_only( ARM, CTRL = c("B: Placebo"), TRT = c("A: Drug X", "C: Combination") ), BMRKR1 = ifelse(BEP01FL == "N", NA, BMRKR1) ) %>% var_relabel( BEP01FL = "BEP" )```## PlotWe then perform with the `fit_survival_step()` function the required calculations: this function fits the Subgroup Treatment Effect Pattern (STEP) models for the survival outcome within each of the percentile intervals of the biomarker variable defining the subgroups.The treatment arm variable must have exactly two levels, where the first one is taken as reference, i.e. the estimated hazard ratios are for the comparison of the second level vs. the first one.In this example we fit the default model where a constant treatment effect is estimated in each of the subgroups that are created according to biomarker quantiles.```{r}vars <-list(time ="AVAL",event ="is_event",arm ="ARM_BIN",biomarker ="BMRKR1")step_matrix <-fit_survival_step(variables = vars,data = adtte)```In this second example we fit instead a model with quadratic biomarker interaction term and we control the number of points at which the hazard ratios are estimated.```{r}step_matrix <-fit_survival_step(variables = vars,data = adtte,control =c(control_coxph(), control_step(degree =2, num_points =15L)))```Before we can plot the data, the `broom::tidy()` method needs to be applied to the STEP result to obtain graph ready data.Thereafter with the `g_step()` function we can create a graph showing the estimated HR along the continuous biomarker subgroups.```{r}step_data <- broom::tidy(step_matrix)graph <-g_step(step_data)graph```We can also add a reference line.```{r}graph +geom_hline(aes(yintercept =1), linetype =2)```{{< include ../misc/session_info.qmd >}}:::